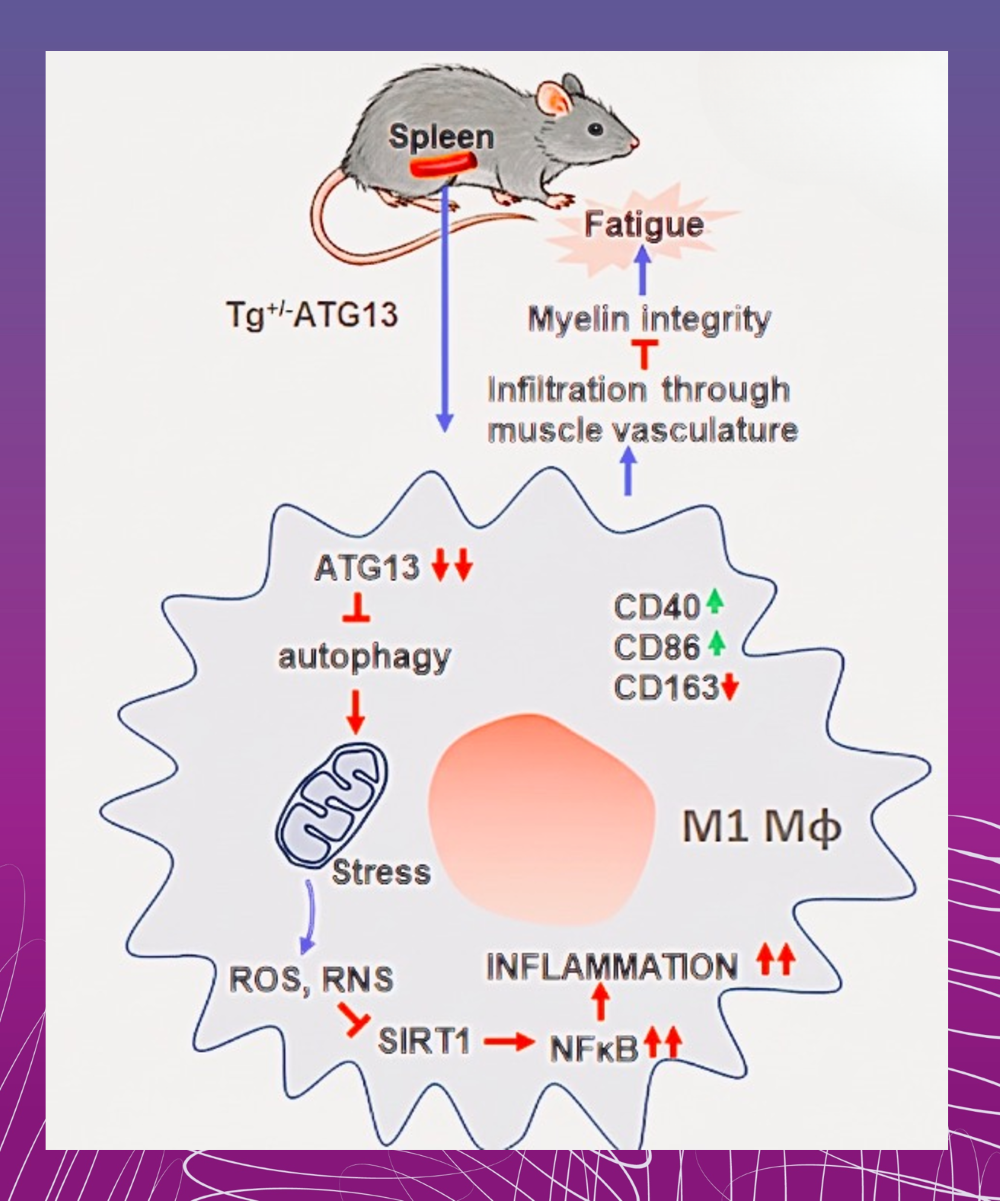
Explaining the Molecular Mechanism Behind PEM Findings: In our current publication, we demonstrate that the genetic depletion of ATG13 gene and the subsequent autophagy impairment may initiate a series of metabolic changes in myeloid cells. Autophagy, which is a necessary clean up process for cells that revitalizes energy metabolism, doesn't work correctly when ATG13 is missing. - The changes we document start with the deficit in mitochondrial oxygen consumption,
- then energy-deficient cells produce inflammatory reactive oxygen species, or oxidative stress,
- then an enzyme called SIRT1 becomes inactive, which neutralizes NF-κB, turning off this anti-inflammatory switch,
- then these series of metabolic changes induce macrophages to take on an inflammatory phenotype, impairing myelin integrity in muscle serving nerves.
- And the inflammatory steps worsen with exercise, as demonstrated in ATG13 deficient mice after time on a treadmill.
Summary: The pathway we describe highlights how NF-kB-mediated inflammatory changes in myeloid cells may contribute to the symptoms of muscle fatigue, post-exertional malaise, and potentially post-infectious fatigue syndromes. Collaborators: University of Wisconsin Milwaukee, Milwaukee Institute of Drug Discovery |
|